pibase | pibase step 4 | Network
Networks are a method to analyze, compare, and visualise complex and not-so-complex data.
A group of samples can be compared pair-wise with respect to a reference sample using pibase_fisherdiff. After these pair-wise comparisons, an rdf file can be generated using pibase_to_rdf (see pibase_to_rdf.sh example). The rdf file can be imported into the phylogenetic Network software for generating and interactively visualizing a median joining network.
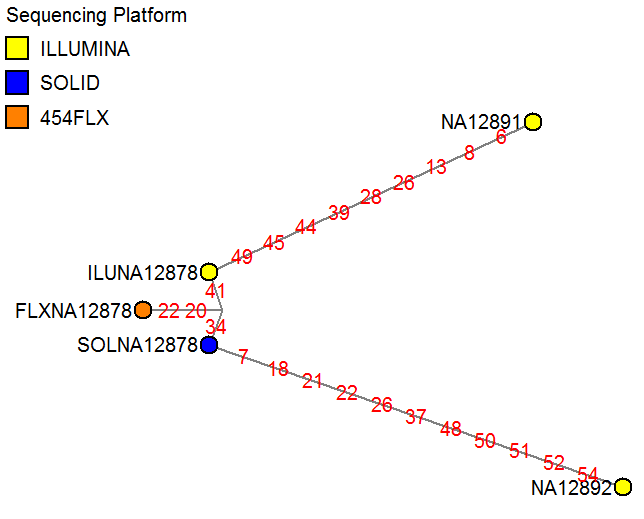
The network links show the IDs of the discriminating SNPs between each sample (details see Forster et al). Networks can be used for re-constructing evolutionary trees, e.g. to identify putative ancestral nodes inferred from a group of known present-day individuals.
The network below shows a sample mix-up test, to check whether the three putative NA12878 sample runs cluster together and not to a different HapMap sample (NA12891 or NA12892). Networks can be used to discriminate between samples using a fairly small amount of known SNPs (here: 55 exonic SNPs in chromosome 22), and can be easier to interpret than cluster plots or identity-by-state tables if larger numbers of samples are analyzed.
Note: If an ancestor needs to be inferred, SNPs from non-recombining loci should be used. When analyzing a group of individuals, mtDNA profiles are suitable; otherwise, genomic SNPs from within a single short genomic region (e.g. within a single gene) may be suitable. When analyzing a group of tissue/blood samples (or single cells) from a single individual, mtDNA profiles and/or any genomic SNPs should be suitable.